



Содержание
Перейти к:
https://doi.org/10.21886/2712-8156-2025-6-1-88-94
Перейти к:
Спиноцеребеллярная атаксия — нейродегенеративное заболевание с аутосомно-доминатным типом наследования, быстрым прогрессированием клинических проявлений с дебютом в молодом возрасте. Рассмотрен клинический случай пациентки (34 года) со спиноцеребеллярной атаксией I типа, отягощённой наследственным анамнезом и формированием феномена антиципации. Отмечено, что клинические симптомы опережают данные нейровизуализации.
Фомина-Чертоусова Н.А., Пивачева Е.С., Домрачева А.М., Созаева Д.И. Семейный случай спиноцеребеллярной атаксии I типа. Клинико-диагностические параллели. Южно-Российский журнал терапевтической практики. 2025;6(1):88-94. https://doi.org/10.21886/2712-8156-2025-6-1-88-94
Fomina-Chertousova N.A., Pivacheva E.S., Domracheva A.M., Sozaeva D.I. A familial case of spinocerebellar attack type I. Clinical and diagnostic parallels. South Russian Journal of Therapeutic Practice. 2025;6(1):88-94. (In Russ.) https://doi.org/10.21886/2712-8156-2025-6-1-88-94
Спиноцеребеллярная атаксия 1 типа (СЦА 1 типа) — это прогрессирующее нейродегенеративное заболевание, вызванное экспансией нуклеотидных последовательностей (CAG-повторов) в 8-м экзоне гена ATXN1 с аутосомно-доминантным типом наследования [1].
Эпидемиология заболевания в среднем составляет 1–2 случая на 100 000 человек, однако существуют регионы с более высоким уровнем заболеваемости. Во всём мире СЦА 1 типа выявляется примерно у 6% пациентов с аутосомно-доминантной мозжечковой атаксией. Однако этот показатель значительно варьируется в зависимости от этнической принадлежности и географического региона. Согласно исследованию D. K. Jin et.all., СЦА 1 типа не была выявлена среди пациентов Южной Кореи, страдающих аутосомно-доминантной атаксией, в то время как в Восточной Сибири, по данным Платонова Ф.А. и соавт., СЦА 1 типа в 100% случаях выявляется у пациентов с аутосомно-доминантной атаксией [2][3][4][5]. В Якутии из-за длительной географической и относительной этнической изолированности заболеваемость наследственной мозжечковой атаксией среди популяции якутов достигла 38,6 на 100 тыс. населения в связи с чем высокий уровень распространённости болезни был оценен как «сибирский очаг» накопления СЦА 1 типа — крупнейший в мире.
Причиной развития СЦА 1 типа является увеличение CAG-повторов в гене ATXN1 локуса 6p22.3. CAG-повторы — это участки ДНК с многократно повторяющимися триплетами нуклеотидов (цитозин-аденин-гуанин). Участки ДНК с непрерывными CAG-повторами содержат в одном или в нескольких местах перерывы вставками триплета нуклеотидов цитозин-аденин-тимин так называемые CAT-перерывы. В норме количество CAG-повторов варьируется в пределах от 6 до 38 с 1–3 САТ-перерывами, которые, как предполагается, участвуют в поддержании стабильности тринуклеотидного участка во время репликации ДНК. Симптомы заболевания выявляются при наличии 39 и более CAG-повторов.
Клеточные механизмы патогенеза СЦА 1 типа обусловлены наличием мутантного белка атаксина-1. Белок экспрессируется в нейронах и клетках глии ЦНС, в мозжечке атаксин-1 локализован как в цитоплазме, так и в ядре клеток Пуркинье. Увеличение CAG-повторов в гене ATXN1 приводит к экспансии полиглутаминового тракта атаксина-1, вызывая тем самым полиглутаминовый механизм приобретения новых нейротоксических функции, приводящий к развитию СЦА 1 типа.
Существует прямая взаимосвязь между количеством CAG-повторов и тяжестью заболевания (чем больше CAG-повторов, тем тяжелее оно протекает). Также существует обратная взаимосвязь между количеством CAG-повторов и дебютом заболевания (чем меньше CAG-повторов, тем позже дебютирует заболевание). У пациентов с более чем 52 CAG-повторами заболевание приводило к тяжёлой инвалидизации в первые 5 лет. Пенетрантность СЦА 1 типа составляет более 95% в семьях с ранним дебютом.
Морфологически для СЦА 1 типа характерна дегенерация мозжечка и средних его ножек, ствола мозга с атрофией вентральных отделов моста, гибель нейронов нижней оливы, двигательных нейронов спинного мозга и периферической нервной системы. Помимо этого наблюдается демиелинизация и ранняя активация астроцитов и микроглии [6].
Дебют СЦА 1 типа приходится на третье-четвёртое десятилетия жизни, однако первые проявления заболевания могут отмечаться в возрастном промежутке от 4 до 74 лет. Встречаются случаи с дебютом после 60 лет, при этом отмечается чисто мозжечковый фенотип. В детском возрасте (до 13 лет) наблюдается более быстрое прогрессирование и более тяжёлое течение заболевания. Период от появления первых симптомов болезни до смерти варьируется от 10 до 30 лет и в среднем составляет около 15 лет. При ювенильной форме тяжёлое поражение ствола мозга приводит к смерти больного в течение 4–8 лет после появления первых клинических симптомов. Для СЦА 1 типа характерен феномен антиципации, что проявляется в более раннем дебюте заболевания и более тяжёлом течении заболевания у потомков. При СЦА 1 типа экспансия нуклеотидных последовательностей гена ATXN1 чаще происходит при передаче патогенного аллеля по отцовской линии. Это обусловлено преимущественным удлинением мутантного повтора в мужском гаметогенезе. При передаче гена от матери область повтора, как правило, остается стабильной.
Клинически СЦА 1 типа на ранних стадиях проявляется нарушением походки, координации, речи, оживлением сухожильных рефлексов, лёгкой дисфагией (поперхивание твёрдой и жидкой пищей), а также появлением гиперметрических саккад и нистагма [7][8]. Больные впервые замечают нарушения походки, когда сталкиваются с проблемой поддержания равновесия при спуске по лестнице или резких поворотах. Однако спортсмены, чья деятельность связана с такими видами спорта, как катание на лыжах или танцы, требующие высокой точности координации движений, могут заметить первые симптомы на более ранних стадиях заболевания.
По мере прогрессирования заболевания скорость саккад замедляется, развивается паралич взора, а также дисметрия, гипотония и дисдиадохокинез. У некоторых пациентов наблюдается атрофия зрительного нерва, выраженная макулопатия, что указывает на нейродегенеративный процесс. Данные изменения проявляются снижением остроты зрения и нарушением цветового зрения [9][10][11].
На поздних стадиях заболевания характерно развитие атрофии мышц, снижение сухожильных рефлексов, когнитивные нарушения и бульбарная дисфункция. Отмечается атрофия жевательных и мимических мышц, периоральные фасцикуляции, выраженная дисфагия, приводящая к частой аспирации пищей. В конечном итоге развиваются респираторные осложнения, дыхательная недостаточность, что является основной причиной гибели больных.
Спектр когнитивных нарушений при СЦА 1 типа шире, и их развитие происходит быстрее, чем при СЦА других генетических варианов [12][13][14][15]. У больных отмечается наличие тревожного расстройства, депрессии и суицидальных мыслей, снижение внимания и памяти [16][17]. У 82% больных СЦА 1 типа выявляется сенсомоторная, смешанная (аксонально-демиелинизирующая) полинейропатия [18]. Экстрапирамидные синдромы отмечаются реже и выявляются у 37,5% пациентов, включая в себя дистонию, брадикинезию (по 33,3%) и постуральный тремор (26,7%) [19].
При диагностике СЦА I типа важное значение имеет магнитно-резонансная томография (МРТ) церебральных структур при которой определяется атрофия мозжечка и ствола мозга [20]. При электронейромиографии (ЭНМГ) выявляются признаки аксонально- сенсорной нейропатии. Молекулярно-генетическое тестирование проводится для выявления аномальной экспансии CAG-повтора в ATXN1. С помощью метода анализа данных нейровизуализации, позволяющий исследовать фокальные различия в анатомии мозга, используя подход статистического параметрического картирования (воксельной морфометрии) определяется уменьшение объёма серого, белого вещества мозжечка, ствола мозга и атрофия спинного мозга [21][22][23][24].
Дифференциальная диагностика СЦА 1 типа проводится с условно курабельными атаксиями различного происхождения, обусловленными такими причинами, как болезнь Вильсона-Коновалова, дефицит витамина В12 и ряда других ферментов, гипотиреоз, паранеопластический синдром. Аутосомно-рецессивные атаксии (атаксия Фридрейха, митохондриальные атаксии и атаксия-телеангиэктазия) клинически сложно отличить от спиноцеребеллярной атаксии, так как их клинические проявления достаточно схожи, что требует проведение молекулярно-генетического исследования. Важно помнить о синдроме Герстманна-Штраусслера-Шейнкера — это прионное заболевание с аутосомно-доминантным типом наследования и дебютом на третьем-четвёртом десятилетии жизни, проявляющееся мозжечковой атаксией, дизартрией, нистагмом, мимикрирующее под спиноцеребеллярную атаксию. Для подтверждения диагноза необходимо проведение молекулярно-генетического анализа гена PRNP (Prion-related protein — белок, связанный с прионом), кодирующего нормальный белок (PrPC) и изоформу этого белка (прионный белок PrPSc с аномальной третичной структурой, участвующий в развитии заболевания). Также дифференциальная диагностика проводится с эпизодической атаксией — группой заболеваний с аутосомно-доминантным типом наследования, провоцирующейся определёнными факторами, такими как эмоциональный и физический стресс, и проявляющейся повторными острыми приступами нарушения координации различной длительности нередко в сочетании с мозжечковыми симптомами [25].
Пациентка Т., 1989 г. р. (34 года), с 2022 г. наблюдается в Неврологическом центре клиники ФГБОУ ВО РостГМУ Минздрава России. При поступлении предъявляла жалобы на шаткость и неустойчивость при ходьбе (походка с широко расставленными ногами), периодическое нарушение глотания жидкой пищи, нарушение речи (отсутствие плавности звукопроизношения, дрожание голоса), координации движений, изменение почерка, эпизоды потемнения в глазах, снижение зрения на оба глаза, периодические крампи в проксимальных отделах ног, императивные позывы на мочеиспускание.
Анамнез заболевания. Дебют заболевания в 29 лет (2018 г.), когда пациентка впервые стала отмечать появление шаткости при ходьбе. В возрасте 32 лет (2021 г.) отмечено изменение речи и усиление неустойчивости при ходьбе.
Наследственный анамнез. У отца, выросшего в детском доме (родители неизвестны), в возрасте 32 лет отмечалась шаткость при ходьбе (умер от ожоговой болезни в 40 лет). Аналогичные симптомы наблюдались у старшей сестры пациентки 1981 г. р., с дебютом в 24 года. Установлен диагноз «Рассеянный склероз». Женщина умерла в 30 лет, причина смерти неизвестна. У родной племянницы (дочь старшей сестры) 2002 г. р. появились изменения речи в 2022 г. Сын пациентки Т. (2009 г. р.) клинически здоров.
На основании представленных анамнестических данных нами была составлена родословная пациентки Т. (рис. 1).
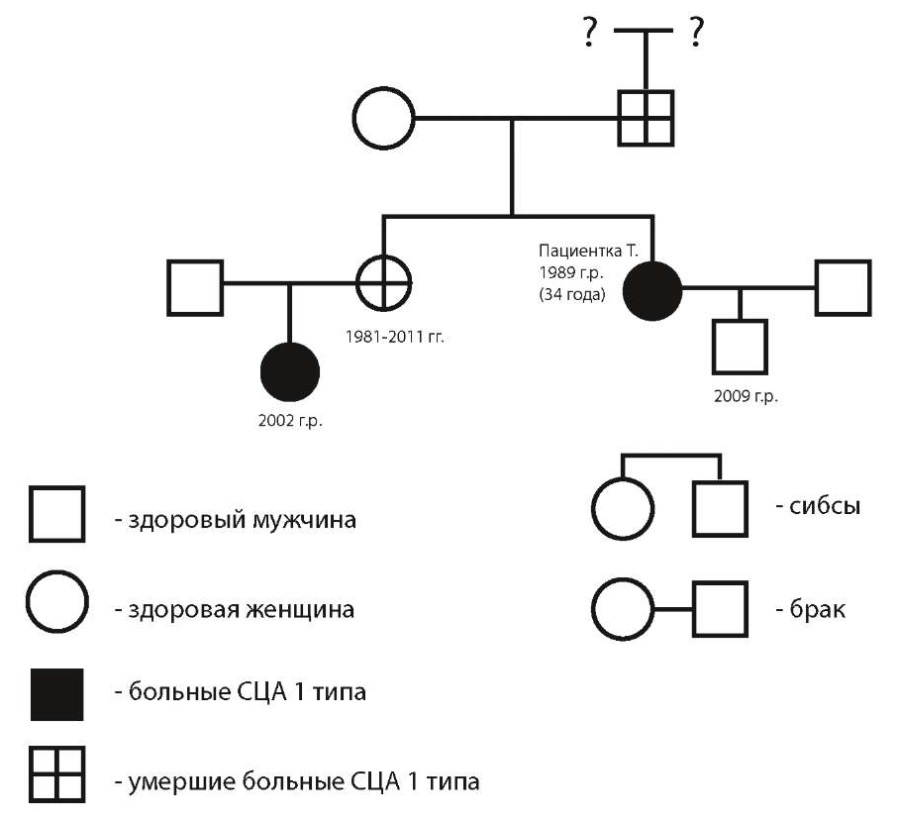
Рисунок 1. Родословная пациентки Т.
Figure 1. Pedigree of patient T.
При оценке неврологического статуса выявлено следующее: сознание ясное, ориентация в пространстве и во времени, в собственной личности не нарушена. ЧН: обоняние не нарушено, зрачки D=S, фотореакция живая, ограничения полей зрения контрольно-сравнительным способом не выявлено, двоение вверх и влево. Ретракция век, двусторонний экзофтальм, парез взора вверх, саккады. Аккомодация и конвергенция не нарушены. Тригеминальные точки при пальпации безболезненны, чувствительность на лице не нарушена. Мимика симметричная, асимметрии носогубных складок нет, язык по средней линии, гнусавый оттенок голоса, речь изменена по типу мозжечковой дизартрии, глотание и вкус не нарушены. Шепотную речь различает, слева и справа на расстоянии 6 метров. Функции грудинно-ключично-сосцевидной и трапециевидной мышц не нарушены. Двигательная сфера: походка с широко расставленными ногами по типу мозжечковой атаксии. Мышечный тонус: D=S, повышение тонуса по экстрапирамидному типу (феномен «зубчатого колеса», пластический тонус). Сухожильные рефлексы с верхних и нижних конечностей: D=S, высокие, с расширением рефлексогенных зон. Патологические стопные знаки (+) с двух сторон. В позе Ромберга выраженная шаткость без сторонности, усиливающаяся при закрывании глаз. Пальценосовая и пяточно-коленная пробы выполняет с легкой интенцией S>D и мимопопаданием. Нарушение глубокой чувствительности в пальцах ног и рук. Нарушение поверхностной чувствительности объективно не выявлено. Тазовые функции: императивные позывы на мочеиспускание. По Монреальской шкале оценки когнитивных функций (МоСА), 28 баллов (соответствует норме) и по госпитальной шкале тревоги и депрессии (HADS): сумма Т-5 (соответствует норме), сумма Д-3 (соответствует норме).
Проведено МРТ головного мозга (2019 г.), не выявившее признаков патологических изменений структур головного мозга. Повторное МРТ головного мозга (2022 г.) выявило МР-признаки существенных атрофических изменений моста, продолговатого мозга и мозжечка. При офтальмологическом обследовании (2022 г.) диагностирована миопия слабой степени, сложный миопический астигматизм с периферической хориоретинальной дистрофией обоих глаз. При проведении стимуляционной ЭНМГ (2022 г.) обнаружены признаки генерализованного симметричного сенсо-моторного поражения периферических нервов верхних и нижних конечностей по демиелинизирующему типу с вовлечением аксонов сенсорных нервов.
Результаты молекулярно-генетического исследования (ДНК-анализа, 2023 г.) представлены в таблице 1.
Таблица / Table 1
Молекулярно-генетическое исследование при спиноцеребеллярной атаксии 1, 2, 3 типа (количество тринуклеотидных повторов (CAG) гена ATXN1 (атаксин 1), ATXN2 (атаксин 2), ATXN3 (атаксин 3)
Molecular genetic study for spinocerebellar ataxia types 1, 2, 3 (number of trinucleotide repeats (CAG) gene ATXN1 (ataxin 1), ATXN2 (ataxin 2), ATXN3 (ataxin 3)
Количество CAG повторов / Number of CAG repeats | ||
Количество CAG повторов в гене ATXN1 Number of CAG repeats in the ATXN1 gene | Количество CAG повторов в гене ATXN2 Number of CAG repeats in the ATXN2 gene | Количество CAG повторов в гене ATXN3 Number of CAG repeats in the ATXN3 gene |
n1 = 28 n2 = 51 | n1 < 35 n2 < 35 | n1 < 53 n2 < 53 |
Примечание: n1 — первая аллель гена, n2 — вторая аллель гена.
Note: n1 — the first allele of the gene, n2 — second allele of the gene.
В результате анализа ДНК пациентки Т. выявлено нормальное число копий CAG-повторов, локализованных в генах ATXN2, ATXN3. Вместе с тем в одной из хромосом выявлено увеличенное число копий CAG-повтора, локализованного в гене ATXN1.
Заключительный клинический диагноз: СЦА 1 типа (ATXN1), спастическая форма, аутосомно-доминантный тип наследования, смешанная атаксия (мозжечковая, заднестолбовая), с нарушением функции тазовых органов, парезом взора вверх, нарушением функции ходьбы. Код МКБ-10: G11.8, G25.8
С учётом полученных данных для племянницы пациентки Е. было выполнено молекулярно-генетическое исследование. В результате анализа ДНК выявлено нормальное число копий CAG-повторов, локализованных в генах ATXN2, ATXN3, в одной из хромосом обнаружено увеличенное число копий CAG-повтора, локализованного в гене ATXN 1. Установлен диагноз «Спиноцеребеллярная атаксия (ATXN1), увеличение CAG повторов до 53, аутосомно-доминантный тип наследования» (табл. 2).
Таблица / Table 2
Молекулярно-генетическое исследование при спиноцеребеллярной атаксии 1, 2, 3 типа (количество тринуклеотидных повторов (CAG) гена ATXN1 (атаксин 1), ATXN2 (атаксин 2), ATXN3 (атаксин 3)
Molecular genetic study for spinocerebellar ataxia types 1, 2, 3 (number of trinucleotide repeats (CAG) of the ATXN1 (ataxin 1), ATXN2 (ataxin 2), ATXN3 (ataxin 3)
Количество CAG повторов / Number of CAG repeats | ||
Количество CAG повторов в гене ATXN1 Number of CAG repeats in the ATXN1 gene | Количество CAG повторов в гене ATXN2 Number of CAG repeats in the ATXN2 gene | Количество CAG повторов в гене ATXN3 Number of CAG repeats in the ATXN3 gene |
n1 = 28 n2 = 53 | n1 < 35 n2 < 35 | n1 < 53 n2 < 53 |
Примечание: n1 — первая аллель гена, n2 — вторая аллель гена.
Note: n1 — the first allele of the gene, n2 — second allele of the gene.
С учётом высокой пенетрантности и аутосомно-доминантного типа наследования СЦА 1 типа при составлении генеалогического древа, как правило, выявляется отягощённый семейный анамнез.
Для СЦА 1 типа характерен феномен антиципации, как и для многих болезней экспансии тринуклеотидных повторов. В данном семейном случае отмечена передача гена от отца двум дочерям, с более ранним дебютом заболевания.
При изучении клинико-диагностических параллелей отмечено, что клинические симптомы могут опережать данные нейровизуализации.
Подтверждён феномен тяжёлой инвалидизации и гибели пациентов в семье у лиц с ранней формой СЦА 1 типа (продолжительность жизни старшей сестры пациентки Т. — 6 лет).
Доказана обратная связь между числом тринуклеотидных повторов и дебютом заболевания.
1. Kerkhof LMC, van de Warrenburg BPC, van Roon-Mom WMC, Buijsen RAM. Therapeutic Strategies for Spinocerebellar Ataxia Type 1. Biomolecules. 2023;13(5):788. DOI: 10.3390/biom13050788
2. Jin DK, Oh MR, Song SM, Koh SW, Lee M, Kim GM, et al. Frequency of spinocerebellar ataxia types 1,2,3,6,7 and dentatorubral pallidoluysian atrophy mutations in Korean patients with spinocerebellar ataxia. J Neurol. 1999;246(3):207-210. DOI: 10.1007/s004150050335
3. Тихонов Д.Г., Гольдфарб Л.Г., Неустроева Т.С., Яковлева Н.В., Тимофеев Л.Ф., Луцкан И.П., и др. Анализ продолжительности жизни и смертности больных спиноцеребеллярной атаксией 1 типа. Проблемы социальной гигиены, здравоохранения и истории медицины. 2015;23(6):31-34. eLIBRARY ID: 25304718 EDN: VHTIDV
4. Opal P., Ashizawa T. Spinocerebellar Ataxia Type 1. In: Adam M.P., Feldman J., Mirzaa G.M., eds. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2025.
5. Platonov FA, Tyryshkin K, Tikhonov DG, Neustroyeva TS, Sivtseva TM, Yakovleva NV, et al. Genetic fitness and selection intensity in a population affected with high-incidence spinocerebellar ataxia type 1. Neurogenetics. 2016;17(3):179-185. DOI: 10.1007/s10048-016-0481-5
6. Tejwani L, Lim J. Pathogenic mechanisms underlying spinocerebellar ataxia type 1. Cell Mol Life Sci. 2020;77(20):4015-4029. DOI: 10.1007/s00018-020-03520-z
7. Donato SD, Mariotti C, Taroni F. Spinocerebellar ataxia type 1. Handb Clin Neurol. 2012;103:399-421. DOI: 10.1016/B978-0-444-51892-7.00025-5
8. Matilla-Dueñas A, Goold R, Giunti P. Clinical, genetic, molecular, and pathophysiological insights into spinocerebellar ataxia type 1. Cerebellum. 2008;7(2):106-114. DOI: 10.1007/s12311-008-0009-0
9. Lebranchu P, Le Meur G, Magot A, David A, Verny C, Weber M, et al. Maculopathy and spinocerebellar ataxia type 1: a new association? J Neuroophthalmol. 2013;33(3):225-231. DOI: 10.1097/WNO.0b013e31828d4add
10. Oertel FC, Zeitz O, Rönnefarth M, Bereuter C, Motamedi S, Zimmermann HG, et al. Functionally Relevant Maculopathy and Optic Atrophy in Spinocerebellar Ataxia Type 1. Mov Disord Clin Pract. 2020;7(5):502-508. DOI: 10.1002/mdc3.12949
11. Vaclavik V, Borruat FX, Ambresin A, Munier FL. Novel maculopathy in patients with spinocerebellar ataxia type 1 autofluorescence findings and functional characteristics. JAMA Ophthalmol. 2013;131(4):536-538. DOI: 10.1001/jamaophthalmol.2013.1127
12. Fancellu R, Paridi D, Tomasello C, Panzeri M, Castaldo A, Genitrini S, et al. Longitudinal study of cognitive and psychiatric functions in spinocerebellar ataxia types 1 and 2. J Neurol. 2013;260(12):3134-3143. DOI: 10.1007/s00415-013-7138-1
13. Klinke I, Minnerop M, Schmitz-Hübsch T, Hendriks M, Klockgether T, Wüllner U, et al. Neuropsychological features of patients with spinocerebellar ataxia (SCA) types 1, 2, 3, and 6. Cerebellum. 2010;9(3):433-442. DOI: 10.1007/s12311-010-0183-8
14. Ma J, Wu C, Lei J, Zhang X. Cognitive impairments in patients with spinocerebellar ataxia types 1, 2 and 3 are positively correlated to the clinical severity of ataxia symptoms. Int J Clin Exp Med. 2014;7(12):5765-5771. PMID: 25664104; PMCID: PMC4307551.
15. Moriarty A, Cook A, Hunt H, Adams ME, Cipolotti L, Giunti P. A longitudinal investigation into cognition and disease progression in spinocerebellar ataxia types 1, 2, 3, 6, and 7. Orphanet J Rare Dis. 2016;11(1):82. DOI: 10.1186/s13023-016-0447-6
16. Olmos V, Gogia N, Luttik K, Haidery F, Lim J. The extra-cerebellar effects of spinocerebellar ataxia type 1 (SCA1): looking beyond the cerebellum. Cell Mol Life Sci. 2022;79(8):404. DOI: 10.1007/s00018-022-04419-7
17. Tichanek F, Salomova M, Jedlicka J, Kuncova J, Pitule P, Macanova T, et al. Hippocampal mitochondrial dysfunction and psychiatric-relevant behavioral deficits in spinocerebellar ataxia 1 mouse model. Sci Rep. 2020;10(1):5418. DOI: 10.1038/s41598-020-62308-0
18. Linnemann C, Tezenas du Montcel S, Rakowicz M, Schmitz-Hübsch T, Szymanski S, Berciano J, et al. Peripheral Neuropathy in Spinocerebellar Ataxia Type 1, 2, 3, and 6. Cerebellum. 2016;15(2):165-173. DOI: 10.1007/s12311-015-0684-6
19. Jhunjhunwala K, Netravathi M, Purushottam M, Jain S, Pal PK. Profile of extrapyramidal manifestations in 85 patients with spinocerebellar ataxia type 1, 2 and 3. J Clin Neurosci. 2014;21(6):1002-1006. DOI: 10.1016/j.jocn.2013.10.021
20. Döhlinger S, Hauser TK, Borkert J, Luft AR, Schulz JB. Magnetic resonance imaging in spinocerebellar ataxias. Cerebellum. 2008;7(2):204-214. DOI: 10.1007/s12311-008-0025-0
21. Ginestroni A, Della Nave R, Tessa C, Giannelli M, De Grandis D, Plasmati R, Salvi F, Piacentini S, Mascalchi M. Brain structural damage in spinocerebellar ataxia type 1 : a VBM study. J Neurol. 2008;255(8):1153-1158. DOI: 10.1007/s00415-008-0860-4
22. Goel G, Pal PK, Ravishankar S, Venkatasubramanian G, Jayakumar PN, Krishna N, et al. Gray matter volume deficits in spinocerebellar ataxia: an optimized voxel based morphometric study. Parkinsonism Relat Disord. 2011;17(7):521-527. DOI: 10.1016/j.parkreldis.2011.04.008
23. Guerrini L, Lolli F, Ginestroni A, Belli G, Della Nave R, Tessa C, et al. Brainstem neurodegeneration correlates with clinical dysfunction in SCA1 but not in SCA2. A quantitative volumetric, diffusion and proton spectroscopy MR study. Brain. 2004;127(Pt 8):1785-1795. DOI: 10.1093/brain/awh201
24. Pedroso JL, Barsottini OG. Spinal cord atrophy in spinocerebellar ataxia type 1. Arq Neuropsiquiatr. 2013;71(12):977. DOI: 10.1590/0004-282X20130187
25. Bhandari J., Thada P.K., Samanta D. Spinocerebellar Ataxia. Treasure Island (FL): StatPearls Publishing; 2023.
Неонила Анатольевна Фомина-Чертоусова, к. м. н., доцент
кафедра нервных болезней и нейрохирургии
Ростов-на-Дону
Елена Сергеевна Пивачева, ординатор 2-го года обучения
кафедра нервных болезней и нейрохирургии
Ростов-на-Дону
Анастасия Михайловна Домрачева, врач-невролог
неврологическое отделение № 1
Ростов-на-Дону
Диана Измаиловна Созаева, д. м. н., доцент
кафедра неврологии, восстановительной медицины и остеопатии
Ростов-на-Дону
Фомина-Чертоусова Н.А., Пивачева Е.С., Домрачева А.М., Созаева Д.И. Семейный случай спиноцеребеллярной атаксии I типа. Клинико-диагностические параллели. Южно-Российский журнал терапевтической практики. 2025;6(1):88-94. https://doi.org/10.21886/2712-8156-2025-6-1-88-94
Fomina-Chertousova N.A., Pivacheva E.S., Domracheva A.M., Sozaeva D.I. A familial case of spinocerebellar attack type I. Clinical and diagnostic parallels. South Russian Journal of Therapeutic Practice. 2025;6(1):88-94. (In Russ.) https://doi.org/10.21886/2712-8156-2025-6-1-88-94
344022, г. Ростов-на-Дону, пер. Нахичеванский, 29
tel.: +79185710558
e-mail: therapeuticjour@gmail.com (sylanadel@gmail.com)






















