



Содержание
Перейти к:
https://doi.org/10.21886/2712-8156-2025-6-2-118-128
Перейти к:
Несмотря на гетерогенную этиологию миелопатий и стереотипность основных клинических проявлений поражений спинного мозга, патогномоничные клинические особенности, признаки вовлечения экстраспинальных структур, особенности течения патологического процесса, различия представления очагов поражения на сагиттальных и аксиальных магнитно-резонансных изображениях и данные лабораторной диагностики позволяют проводить дифференциальную диагностику как между воспалительными и невоспалительными формами, так и внутри самих этих групп.
Сорокин Ю.Н., Сорокина Е.Ю. Особенности дифференциальной диагностики воспалительных и невоспалительных миелопатий. Южно-Российский журнал терапевтической практики. 2025;6(2):118-128. https://doi.org/10.21886/2712-8156-2025-6-2-118-128
Sorokin Yu.N., Sorokina E.Yu. Peculiarities of differential diagnostics of inflammatory and noninflammatory myelopathies. South Russian Journal of Therapeutic Practice. 2025;6(2):118-128. (In Russ.) https://doi.org/10.21886/2712-8156-2025-6-2-118-128
Изолированные поражения спинного мозга в клинической практике встречаются относительно нечасто, при этом в этиологическом отношении миелопатии представляют собой гетерогенную группу заболеваний (табл.), что требует проведения широкой дифференциальной диагностики не только между воспалительными и невоспалительными формами, но и внутри самих этих групп [1–3]. В случае невозможности установления конкретной причины миелопатия расценивается как идиопатическая.
Таблица / Table
Этиологическая классификация миелопатий [1–3]
Etiological classification of myelopathies [1–3]

Примечание: AQP4+ — аквапорин-4-положительный; GFAP — glial fibrillary acidic protein; MOG — myelin oligodendrocyte glycoprotein; TNF-α — фактор некроза опухоли-α.
Note: AQP4+ — aquaporin-4-positive; GFAP — glial fibrillary acidic protein; MOG — myelin oligodendrocyte glycoprotein; TNF-α — tumor necrosis factor-α
Несмотря на широкий спектр этиологии, основные клинические проявления собственно поражения спинного мозга достаточно стереотипны. В зависимости от уровня поражения развиваются симметричные или асимметричные спастические или вялые парезы/параличи конечностей (верхних и/или нижних), сенсорные расстройства сегментарного или проводникового типа, в том числе сенситивная атаксия, при двустороннем поражении — дисфункция мочевого пузыря или кишечника и сексуальная дисфункция. При этом избирательное поражение белого вещества может быть признаком рассеянного склероза (РС), подострой комбинированной дегенерации, паранеопластической миелопатии и радиационного миелита, а преимущественное поражение серого вещества может быть проявлением инфаркта спинного мозга, вирусного миелита или MOG-ассоциированного заболевания [4][5].
Вместе с тем некоторые заболевания, помимо типичной симптоматики миелопатии, имеют достаточно патогномоничные клинические особенности или признаки вовлечения экстраспинальных структур. Например, диагностике болезни Бехчета способствует выявление сопутствующих системных проявлений, которые включают наличие язв в полости рта или на половых органах, дерматологические изменения (папуло-пустулезные поражения, узловатая эритема, патергическая реакция), увеит и сосудистые поражения (флебит, артериальный тромбоз, тромбоз крупных вен, аневризма).
Для AQP4+ расстройств спектра оптиконейромиелита (AQP4+ РСОНМ) характерными симптомами являются некупируемые в течение дней или нескольких недель тошнота, рвота, непреодолимая икота (за счёт вовлечения «самого заднего поля — области area postrema в дорсальных отделах продолговатого мозга), нейропатические боль, зуд или развитие болезненных пароксизмальных тонических мышечных спазмов (короткие, длительностью 15–60 сек., эпизоды непроизвольных сокращений сгибателей и разгибателей конечностей или мышц туловища несколько раз в день, иногда провоцируемые движением,), а для MOG-ассоциированных расстройств (MOG-АР) — вялые парезы с арефлексией и стойкое нарушение функции тазовых органов (за счёт вовлечения конуса спинного мозга). В целом фенотип MOG-АР намного шире, чем фенотип AQP4+ РСОНМ, при этом миелит является основным клиническим признаком. При AQP4+ РСОНМ могут наблюдаться клинические проявления, предполагающие демиелинизацию, — феномен Утхоффа и симптом Лермитта (как и при РС), а также (изредка) симптомы вовлечения конуса спинного мозга [3].
Поражение зрительного нерва со снижением остроты зрения и болью в глазах, усиливающейся при движениях глаз, в начале заболевания чаще отмечается при MOG-АР (50-65%) по сравнению с AQP4+ РСОНМ (35%), при этом двустороннее одновременное поражение зрительных нервов часто встречается в обоих случаях — 5–84% и 17–82% соответственно [6]. Для MOG-АР характерно наличие боли в глазах до начала потери зрения, отёк диска зрительного нерва, полное или почти полное восстановление остроты зрения в последующем и отсутствие вовлечения хиазмы, в отличие от AQP4+ РСОНМ.
При GFAP-астроцитопатии изолированный миелит встречается только в 4–5% случаев, как основной синдром — в 11%, а как одно из проявлений менингоэнцефаломиелита — у 30% пациентов. При этом миелиту часто сопутствует папиллит диска зрительного нерва, клинически проявляющийся нечёткостью зрения [3][7].
Одним из основных имитаторов воспалительных миелопатий является инфаркт спинного мозга. В 72% при спинальном инфаркте отмечается интенсивная боль в спине или в конечностях, что связывают с ирритацией спиноталамического тракта или с патогенетическим механизмом развития (фиброзно-хрящевая эмболия, расслоение артерии) [8][9]. Важное значение имеют выявление сердечно-сосудистых факторов риска, факт проведения анестезии, медицинских манипуляций на позвоночнике или аорте.
Другими имитаторами воспалительных миелопатий чаще всего являются метаболические нарушения (митохондриальные расстройства, дефицит витамина В12 и меди), спондилогенная миелопатия, спинальная дуральная артериовенозная фистула, интрамедуллярные опухоли спинного мозга (первичные, такие как эпендимома, астроцитома, или метастатические). Невоспалительные миелопатии чаще отмечаются у лиц мужского пола и более старшего возраста (р<0,001 в обоих случаях) [10].
В связи с однотипностью спинальной клинической симптоматики в случае отсутствия экстраспинальных проявлений при проведении дифференциальной диагностики важное значение имеют острота развития и особенности дальнейшего течения патологического процесса, изменения сыворотки крови и ликвора, количество очагов поражения при магнитно-резонансной томографии (МРТ), особенности их продольного распространения и расположения в аксиальной плоскости спинного мозга на МРТ-изображениях, специфические паттерны контрастного усиления и последующая динамика МРТ-изменений [1][2][11].
Одним из наиболее важных факторов дифференциальной диагностики является временнóй профиль развития симптоматики — период от начала заболевания до максимальной выраженности симптомов (надир) [12]. Для иммуноопосредованных или инфекционных миелопатий типично острое или подострое начало заболевания с достижением максимальной выраженности клинической картины в течение 6–48 часов или 2–21 день соответственно. Среди инфекционных миелопатий подострое развитие симптоматики особенно характерно при вирусной этиологии (вирус ветряной оспы, простого герпеса, цитомегаловирус, вирус Западного Нила, вирус иммунодефицита человека (ВИЧ), энтеровирус), реже — при бактериальных или паразитарных возбудителях [3][13][14].
Для вирусов типично поражение преимущественно вещества спинного мозга и нервных корешков или провоцирование развития вторичного иммунного воспаления. Бактериальные, грибковые и паразитарные инфекции характеризуются образованием абсцессов, гранулем и кист, что может сопровождаться вторичной компрессией спинного мозга, напоминая деструктивное поражение при остеомиелите [15].
В целом диагноз «Инфекционное заболевание» должен быть полностью определён на основании клинической картины, анамнестических и лабораторных данных. При этом лихорадка и продромальные системные симптомы не являются специфичными для инфекции и могут выявляться при AQP4+ РСОНМ, активных системных воспалительных заболеваниях, паранеопластических и GFAP-миелопатиях. В то же время, несмотря на достижения мультимодальных методов визуализации и лабораторной диагностики, при подозрении на инфекционную и параинфекционную этиологию миелопатии конкретный возбудитель часто остается неидентифицированным [16][17].
Для иммуноопосредованных миелопатий типична большая вероятность развития рецидивов. При AQP4+ РСОНМ она достигает 90%, при этом рецидивы характеризуются тяжёлой инвалидизацией (в целом выше, чем при РС) и часто неполным восстановлением со ступенчатым накоплением инвалидности. В группе MOG-АР рецидивы реже наблюдаются при фенотипе миелита (16–23%), чем при фенотипах оптического неврита (28–91%) и оптиконейромиелита (50%) Они также проявляются тяжелой инвалидизацией, но восстановление после рецидива обычно полное или почти полное, а долгосрочные результаты часто благоприятны с меньшим накоплением инвалидности. При GFAP-астроцитопатиях рецидивы могут возникать в 20–50% случаев [3].
Рецидивы при РС обычно имеют лёгкую или умеренную степень тяжести и часто являются субклиническими. Клинически проявляющиеся рецидивы в большинстве случаев обычно заканчиваются полным или почти полным клиническим выздоровлением на этапе ремиттирующего течения РС, однако накопление инвалидности отмечается не только во время рецидивов, но и в неактивную фазу заболевания. При этом наличие бессимптомных МРТ-поражений спинного мозга у пациентов с клинически изолированным синдромом ассоциируется с повышенным риском более высокой инвалидности через 5 лет, а у пациентов с радиологически изолированным синдромом — с более высоким риском соответствия диагностическим критериям РС и развития первично-прогрессирующего течения заболевания [18].
Реже острое или подострое начало может быть связано с невоспалительными причинами при дисметаболических миелопатиях, гематомах или мальформациях. Острое или чаще подострое развитие неврологического дефицита с неспецифической симптоматикой, предшествующей болью в спине и явлениями «миелогенной перемежающейся хромоты» характерно для венозных инфарктов спинного мозга, развивающихся при наличии спинальной артериовенозной фистулы твердой мозговой оболочки [8].
Сверхострое начало с развитием максимума выраженности клинической картины в течение менее чем 4–6 часов характерно для некротизирующих паранеопластических миелопатий. При этом альтернативным невоспалительным диагнозом могут быть острая травма или инфаркт спинного мозга, а также метастазы в спинной мозг (наиболее часто развивающиеся из лёгких и молочных желез). В то же время темп развития ишемии спинного мозга может варьироваться от сверхострого и острого до подострого и хронического (в течение нескольких недель). Также сверхостро (в течение несколько часов) может развиться токсическая миелопатия после интраназального введения героина [17][19].
Острые и сверхострые процессы за счёт механизмов спинального шока проявляются низким мышечным тонусом и арефлексией (выявление четкого уровня сенсорных расстройств на туловище должно вызывать подозрение в отношении центрального, а не периферического поражения), а подострые — спастическим повышением мышечного тонуса, гиперрефлексией и патологическими рефлексами.
Хроническое прогрессирующее течение миелопатического синдрома (более 3 недель — месяцы-годы) чаще наблюдается в случае прогредиентного РС и солитарного склероза, реже — при саркоидозе, паранеопластических миелопатиях, болезни Бехчета, GFAP-астроцитопатии. Почти всегда во всех этих случаях миелит сопровождается и поражением головного мозга. При прогредиентном РС прогрессирующее накопление инвалидности отмечается с начала заболевания (первично-прогрессирующий РС) или после первоначально ремиттирующего течения (вторично-прогрессирующий РС), обычно — в виде прогрессирующих двигательных нарушений, что часто совпадает с развитием очаговых атрофических поражений в боковых столбах спинного мозга. Миелопатия может быть начальным проявлением саркоидоза у 79% пациентов с саркоидозом спинного мозга [20].
Невоспалительными имитаторами иммуноопосредованных или инфекционных миелопатий при наличии прогрессирующего течения заболевания могут выступать спондилогенная миелопатия, дуральная артериовенозная фистула, новообразования спинного мозга и поздняя лучевая миелопатия. При этом хроническое развитие симптоматики в этой когорте наблюдается достоверно чаще (р<0,001) по сравнению с группой воспалительных миелопатий [3][10][21].
При любом темпе начала и нарастания неврологической симптоматики — от сверхострого до хронического — в первую очередь необходимо исключить сдавление спинного мозга, причинами которого чаще всего являются дегенеративные заболевания позвоночника. Другими причинами компрессии спинного мозга могут быть травма, эпидуральная патология (метастазы, гематома или абсцесс) и интрадуральные опухоли [17][22].
Особенности спинальной очаговой неврологической симптоматики определяются характером распределения очагов поражения спинного мозга. На основании анатомо-топографической картины и данных нейровизуализации по длиннику спинного мозга различают продольно-распространенный миелит, когда длина очага поражения, измеренная по смежным телам позвонков, составляет 3 и более сегмента в сагиттальной плоскости спинного мозга, и короткий миелит (менее 3 сегментов), а на аксиальных срезах — поперечный миелит, когда очаг поражения по данным МРТ захватывает половину и более поперечника спинного мозга.
Большинство поражений приходится на шейный и грудной сегменты, при некоторых миелопатиях характерно вовлечение и конуса спинного мозга, в связи с чем рекомендуется полная визуализация спинного мозга для выявления множественных поражений и особенностей регионального распределения очагов, а также определение специфических паттернов расположения очагов поражения и накопления контрастного вещества на сагиттальных и аксиальных МРТ-изображениях.
Развитие поперечного миелита наиболее характерно для иммуноопосредованных и инфекционных миелопатий, а также саркоидоза, но возможно и при тотальной ишемии спинного мозга, спинальной артериовенозной фистуле твердой мозговой оболочки, глиомах и спинальных метастазах (в 9% случаев) [3][11].
Сагиттальные изображения спинного мозга демонстрируют одиночные или множественные интрамедуллярные Т2-гиперинтенсивные очаги поражения спинного мозга, которые распространяются вдоль различного числа смежных тел позвонков и вовлекают серое или белое вещество отдельно или в сочетании.
Локализация очагов поражения в шейном и грудном отделах спинного мозга характерна для РС, AQP4+ РСОНМ, MOG-АР, GFAP-астроцитопатии, саркоидоза, болезни Бехчета и паранеопластических миелопатий. Вовлечение конуса спинного мозга наиболее часто отмечается при MOG-АР, реже — при GFAP-астроцитопатии.
При этом невоспалительные миелопатии также характеризуются вовлечением шейных и грудных сегментов, что наблюдается при инфарктах спинного мозга, дисметаболических миелопатиях (обусловленных дефицитом витамина В12 или меди), спинальных эпендимомах, астроцитомах и метастазах. Для спондилогенной миелопатии наиболее характерна шейная локализация, для спинальной дуральной артериовенозной фистулы — грудная. При последней также часто отмечается вовлечение конуса спинного мозга, как и при спинальных эпендимомах и астроцитомах.
Обширные продольные поражения спинного мозга (продольно-распространённый миелит) характерны для большинства иммуноопосредованных миелопатий – AQP4+ РСОНМ, MOG-АР, GFAP-астроцитопатии, саркоидоза, болезни Бехчета, постинфекционных, поствакцинальных и паранеопластических миелопатий, миелопатий при применении ингибиторов иммунных контрольных точек или при системных заболеваниях соединительной ткани, а также возможны как атипичное проявление РС.
При этом для AQP4+ РСОНМ в остром периоде характерны веретенообразное утолщение и отёк спинного мозга, а также яркое пятнистое поражение в пределах продольно обширной зоны Т2-гиперинтенсивности, для MOG-АР — линейное распространение очага поражения в передних отделах спинного мозга («вентральная сагиттальная линия»), а для саркоидоза — линейное распространение очага поражения с вовлечением центрального серого вещества и дорсальных столбов спинного мозга («сагиттальный линейный паттерн») [11].
Поражение спинного мозга на МРТ при GFAP-астроцитопатии отмечается в 30–100% случаев и проявляется едва различимыми, неясными, нечёткими Т2-гиперинтенсивными очагами, менее выраженными и с меньшим отёком спинного мозга, чем при AQP4+ РСОНМ, часто продольно обширными и преимущественно центрально расположенными. В 70% случаев отмечается вовлечение конуса спинного мозга [23].
При паранеопластических миелопатиях выявляются протяжённые линейные продольные Т2-гиперинтенсивные очаги с симметричным поражением боковых или задних столбов, способные накапливать контраст. Подобные тракт-специфические очаги могут встречаться при дефиците витамина В12 или меди, инфекциях (ВИЧ, сифилис) и некоторых наследственных миелопатиях (связанных с геном DARS2), однако в этих случаях накопления контраста не наблюдается. Имитировать паранеопластические нарушения у пациентов с иммуносупрессией также может миелит, связанный с вирусом ветряной оспы.
Короткие поражения спинного мозга (1–2 сегмента) типичны для РС (обычно множественные) и заболеваний спектра РС — прогрессирующего солитарного склероза и чисто рецидивирующего короткого миелита, а как атипичные проявления возможны при AQP4+ РСОНМ (15% как первый эпизод миелита), MOG-АР (38%), саркоидозе и болезни Бехчета. Большинство острых воспалительных поражений сопровождается отёком и накоплением контрастного вещества [3].
Из невоспалительных миелопатий обширные продольные очаги поражения спинного мозга характерны для дефицитарных состояний (витамин В12, медь), спинальной дуральной артериовенозной фистулы, спинальных эпендимом и астроцитом. Инфаркт спинного мозга, спондилогенная миелопатия и спинальные метастазы характеризуются развитием вариабельных по размеру очагов поражения спинного мозга.
При этом инфаркт спинного мозга чаще развивается на грудном уровне, в отличие от воспалительных миелопатий, чаще поражающих шейный отдел, и в передних отделах спинного мозга выявляется Т2-гиперденсивная линейная узкая полоса («знак карандаша»). Подтверждением ишемии спинного мозга может быть сопутствующий инфаркт одного из соседних тел позвонков, однако он обнаруживается только в 4–10% случаев. В первый день очаг ишемии может не выявляться, что обусловливает необходимость проведения повторной МРТ через несколько суток.
Наиболее частая локализация спинальной дуральной артериовенозной фистулы — на уровне между T6-L2 сегментами спинного мозга с частым вовлечением конуса. Протяжённость Т2-гиперинтенсивности не коррелирует со степенью пареза нижних конечностей, сенсорными расстройствами или нарушениями функции сфинктеров. При этом типично увеличение поперечного размера спинного мозга в нижне-грудном отделе, отражающее отёк мозгового вещества, но этот признак всё-таки не является специфическим, поскольку характерен и для других патологических процессов — демиелинизирующих заболеваний, травм и стеноза позвоночного канала вследствие дегенеративных изменений позвоночника. Более специфичный признак — расширенные интрадуральные перимедуллярные вены в виде линейных областей пустоты потока на Т2-изображениях, лучше визуализируемые в режиме миелографии. В случае выраженного отёка спинного мозга вены могут не визуализироваться вследствие масс-эффекта, вследствие чего в половине случаев первоначально выставляется неверный диагноз миелита [11].
Дефицитарные состояния с недостаточностью витамина В12 или меди характеризуются продольно-обширными линейными дорсальными поражениями спинного мозга шейной или грудной локализации, не накапливающими контрастное вещество. Дефицит витамина В12 может имитировать паранеопластические продольно-распространенные миелопатии, Т2-гиперинтенсивные поражения обычно симметричны и ограничиваются задними или латеральными столбами белого вещества. Закись азота деактивирует витамин B12, поэтому её воздействие также может привести к миелопатии с аналогичными признаками, но она более острая, может иногда накапливать контраст, имитируя воспалительную или неопластическую этиологию.
Спинальные эпендимомы часто неправильно диагностируются как AQP4+ РСОНМ. Однако эпендимома визуализируется как Т2-гиперденсивное протяженное образование с локальным расширением спинного мозга и с расположенной рострально или каудально по отношению к Т2-гиперденсивной опухоли Т1-гиподенсивной кистой и формированием вокруг последней Т2-гиподенсивного ободка («знак колпачка»), обусловленного отложением гемосидерина вследствие сопутствующего кровотечения.
Большинство острых поражений при РС и AQP4+ РСОНМ сопровождается кольцевидным накоплением контраста, при этом во втором случае контрастирование может иметь линзовидную или эллипсоидную форму в сагиттальной проекции (продолговатое крылообразное усиление), чего не наблюдается при РС. В то же время при РС возможно и гомогенное контрастирование. Накопление контраста обычно прекращается в течение 3 месяцев. При MOG-АР выявляется вентральное лептоменингеальное накопление контраста, слабое паренхиматозное, кольцевидное отмечается редко.
Саркоидоз во время острой фазы почти у всех пациентов характеризуется лептоменингеальным и линейным дорсальным субпиальным накоплением контраста, которое простирается на 2 и более последовательных позвоночных сегмента в сагиттальной проекции с возможным контрастным усилением и центрального канала. Лептоменингеальное контрастирование также выявляется на уровне конского хвоста и корешков спинномозговых нервов. В то же время в 10% случаев саркоидоз-ассоциированный миелит может развиваться и как передний, с вентральным субпиальным накоплением контраста в непосредственной близости от области дегенерации межпозвонковых дисков в виде косы — с повторяющимися, не накапливающими контраст пробелами на уровне входа передней спинальной артерии («знак косы») [20].
Для паранеопластических поражений также типично дорсальное линейное накопление контраста на сагиттальных срезах, однако аксиальные проекции показывают тракт-специфичное контрастирование белого вещества проводящих путей (боковые и задние столбы спинного мозга) и спинномозговых корешков, а лептоменингеальное накопление контраста не выявляется.
Кроме этого, вследствие развития отёка спинного мозга AQP4+ РСОНМ и саркоидоз часто неправильно диагностируются как злокачественные новообразования, однако для последних характерно как гомогенное, так и неоднородное контрастирование очага поражения, в случаях же метастатического поражения удлинённый кольцевидный паттерн накопления контраста отличается более слабым контрастированием центра области накопления контраста по сравнению с периферией, а верхняя или нижняя часть кольца характеризуется более выраженным контрастированием — появлением «пламени» («знак кольца и пламени»). При этом на аксиальных изображениях в 9% случаев в центре зоны контрастирования отмечается точечное более интенсивное накопление контраста — «знак центральной точки», который не встречается у пациентов с первичными опухолями спинного мозга [11].
При болезни Бехчета лептоменингеальное контрастирование выявляется в 30% случаев, но более специфичны МРТ-изменения на аксиальных изображениях. При GFAP-астроцитопатии накопление контраста может быть точечным (50% случаев), пиальным или лептоменингеальным (40%), может включать центральный канал (30%) или конский хвост (20%) [23].
Невоспалительные миелопатии не во всех случаях сопровождаются накоплением контрастного вещества в очаге поражения. При недостаточности витамина В12 или меди контрастирование очагов не возникает. Для спондилогенной миелопатии характерно накопление контраста в виде поперечной полосы, равной высоте или чуть ниже уровня максимальной компрессии спинного мозга («знак блина»), что отражает очаговое нарушение гемато-спинномозгового барьера.
При инфаркте спинного мозга выявляется линейное накопление контраста в передних отделах соответственно очагу поражения на нативной МРТ («знак карандаша»), а для спинальной дуральной артериовенозной фистулы характерен «знак недостающей части» — очаговое отсутствие накопления контраста в гомогенной обширной области контрастирования, что связано с различиями венозного оттока, при этом расширенные интрадуральные перимедуллярные вены визуализируются в виде извивающихся, змеевидных, накапливающих контраст структур на Т1-изображениях [11].
Аксиальные изображения спинного мозга позволяют изучить характеристики очага поражения и уточнить его локализацию относительно поперечника спинного мозга с выделением 6 паттернов (рис.): центральный (поперечный), периферический, центростремительный, задне-столбовой, избирательное поражение серого вещества, передне-роговой [22].
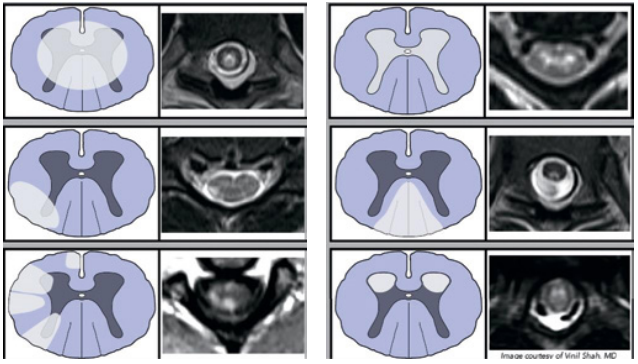
Рисунок. Паттерны поражения поперечника спинного мозга на аксиальных МРТ-изображениях: 1) центральный (поперечный), 2) периферический, 3) центростремительный, 4) задне-столбовой, 5) избирательное поражение серого вещества, 6) передне-роговой [22]
Figure. MRI patterns of the spinal cord axial involvement: 1) central (transverse), 2) peripheral, 3) centripetal, 4) dorsal column, 5) central gray matter, 6) anterior horns [22]
Центральный (поперечный) паттерн: поражение локализовано в центре поперечника спинного мозга и вовлекает половину и более поперечного сечения (поперечный миелит). Классически связан с AQP4+ РСОНМ, MOG-АР, острым рассеянным энцефаломиелитом, системными воспалительными состояниями, инфекционным миелитом, спинальной дуральной артериовенозной фистулой, облучением. Спондилогенная миелопатия на аксиальных изображениях также может проявляться диффузным вовлечением поперечника спинного мозга.
Как и на сагиттальных изображениях, для AQP4+ РСОНМ характерны яркие пятнистые очаги поражения спинного мозга с интенсивностью Т2-сигнала равной или превышающей интенсивность ликвора, отражающие обратимые локальные изменения, связанные с острым воспалением и нарушением гемато-спинномозгового барьера и исчезающие после острой фазы. В то же время такие яркие пятнистые очаги могут обнаруживаться и в 30% случаев инфарктов спинного мозга, что необходимо учитывать при проведении дифференциальной диагностики острых миелопатий.
Периферический паттерн: поражение локализовано в белом веществе по всей окружности поперечника спинного мозга или только в отдельных его столбах. Типичен для РС, при распространении на задние корешки необходимо подозрение в отношении нейроборрелиоза и миелита, ассоциированного с вирусами герпеса. При РС возможно также асимметричное парциальное поражение поперечника спинного мозга за счёт клиновидного вовлечения белого вещества боковых или задних столбов.
Центростремительный паттерн: поражение развивается из лептоменингеальной части и распространяется в паренхиму спинного мозга. Является классическим проявлением саркоидоза, возможен также при туберкулезе и инфильтративных процессах (метастазы, лимфома). Часто связан со стойким (более 3 месяцев) накоплением контраста.
Задне-столбовой паттерн: поражение локализовано в белом веществе задних столбов в виде знака «кроличьи уши» или V-образного шеврона (периферическое тракт-специфичное поражение). Характерен для метаболической и токсической этиологии (дефицит витамина B12 и меди, злоупотребление закисью азота, интоксикация цинком, интратекальное введение метотрексата), паранеопластических состояний, хронических инфекций (спинная сухотка, вакуолярная ВИЧ-миелопатия).
При недостаточности витамина B12 или меди возможно вовлечение и боковых столбов, без накопления контраста. Постинфекционные или поствакцинальные миелопатии также отличаются развитием латеральных или дорсальных трактопатий с нечастым накоплением контраста.
Паттерн избирательного поражения серого вещества: область поражения имеет Н-образную форму («знак Н»). Отмечается при инфаркте спинного мозга, AQP4+ РСОНМ (менее чем в 10% случаев), MOG-АР (в 30% случаев, также могут быть диффузными и необязательно ограничиваются серым веществом, как и при AQP4+ РСОНМ), передней компрессии спинного мозга, дуральной артериовенозной фистуле, остром вялом миелите после энтеровирусных инфекций, энцефаломиелите, вызванном вирусом Западного Нила. Не бывает при РС.
Для миелопатии, связанной с болезнью Бехчета, типичен «знак бублика» — Т2-гиперденсивный очаг почти на все центральное серое вещество в виде кольца с гипоинтенсивным центром (возможно накопление контраста по периферии). Этот признак связывают с нарушением венозного оттока.
При инфаркте спинного мозга в зависимости от полного или неполного поражения бассейна передней спинальной артерии отмечается также вовлечение передних 2/3 поперечника спинного мозга, возможен и синдром монолатерального поражения половины спинного мозга (синдром Броун-Секара ишемического генеза).
Передне-роговой паттерн: поражение локализовано избирательно в сером веществе передних рогов в виде знака «змеиные глаза» или «совиные глаза». Наблюдается при инфаркте спинного мозга, компрессионных, токсических миелопатиях, паранеопластическом миелите, инфекционных миелитах (полиовирус, энтеровирус, вирус Западного Нила). В целом при инфаркте спинного мозга осевая локализация поражения и картина визуализации могут быть различными по длине поражения, один инфаркт может иметь несколько моделей [9].
Как при РС, так и при AQP4+ РСОНМ в 30% случаев наблюдается полное или неполное кольцевидное контрастирование поражений спинного мозга, поэтому его диагностическая ценность в аксиальной проекции для дифференциальной диагностики этих заболеваний ограничена.
При саркоидозе отмечается дорсальное субпиальное накопление контраста, которое обычно расширяется внутрь с контрастированием центрального канала и образованием формы трезубца («знак трезубца»). Накопление контраста остается стойким в течение двух месяцев после начала клинических проявлений более чем у 90% пациентов, сохраняется после шести месяцев и полностью исчезает через 1 год и более.
Паранеопластические миелопатии, в отличие от дисметаболических, демонстрируют тракт-специфическое накопление контраста боковых и задних столбах в 30–50% случаев.
При инфаркте спинного мозга накопление контраста выявляется в подостром периоде в передних рогах («симптом совы или змеиного глаза» или «знак Н»), которые возможны также при AQP4+ РСОНМ. При более выраженной ишемии выявляется более обширное гомогенное контрастирование соответственно зоне кровоснабжения спинальной артерии.
Для спинальной дуральной артериовенозной фистулы характерно однородное накопление контраста на аксиальных изображениях, гомогенное центральное контрастирование расширенных интрадуральных вен, которые обычно расположены на дорсальной поверхности спинного мозга, однако в случае шейной локализации могут располагаться и вентрально.
Спондилогенная миелопатия на аксиальных изображениях характеризуется контрастированием периферической области поперечника спинного мозга и выглядит как окружность, вовлекающая периферическое белое вещество и не затрагивающая центральное серое вещество. После хирургической декомпрессии способность накапливать контраст со временем уменьшается, но полное исчезновение может занять от нескольких месяцев до нескольких лет [11].
Причина поражения спинного мозга после окончания острой фазы всё-таки может остаться неизвестной даже при адекватной нейровизуализации и достаточном лабораторном обследовании. В связи с этим необходимо проведение повторных МРТ-исследований для постановки правильного диагноза и понимания патогенеза заболевания.
При AQP4+ РСОНМ у трети пациентов в течение трёх месяцев продольно обширные поражения подвергаются фрагментации на множественные короткие очаги, которые через 6 и более месяцев от начала заболевания уменьшаются в размерах с возможным разрешением в последующем, но с развитием атрофии спинного мозга вплоть до выраженной в отдалённом периоде. Новые очаги или увеличение бессимптомных Т2-поражений наблюдаются редко (3–13% случаев), однако в целом риск рецидива поле первого клинического эпизода является очень высоким (70–80%) даже после 5 лет ремиссии [3][6].
Для MOG-АР при последующем наблюдении в течение 3–6 и более месяцев характерно полное исчезновение большинства T2-очагов (67–79% случаев), что объясняет отсутствие вторичного прогрессирующего течения и в целом даёт хороший долгосрочный прогноз. Появление новых очагов или увеличение бессимптомных Т2-поражений является также редким (3–14% случаев), как и частота клинических рецидивов (3%) [6].
При РС с наступлением ремиссии типично уменьшение размеров Т2-гиперденсивных очагов в спинном мозге и прекращение накопления контраста, но полностью они редко исчезают. Выявляется очаговая атрофия спинного мозга в местах предшествующей воспалительной демиелинизации. Для РС рецидивирование является характерным, особенно при применении лекарственных средств низкой или умеренной эффективности.
GFAP-астроцитопатия, болезнь Бехчета и саркоидоз также характеризуются уменьшением спинальных очагов с возможным развитием очаговой атрофии спинного мозга. В то же время в 20–50% случаев GFAP-астроцитопатии наблюдается рецидивирующее течение заболевания. У некоторых пациентов с болезнью Бехчета отмечается полное исчезновение очагов.
Острое или подострое развитие синдрома поражения спинного мозга требует проведения всего доступного спектра лабораторной диагностики инфекционного, аутоиммунного, паранеопластического или дисметаболического процесса. При хроническом развитии миелопатии необходимо исключение прогредиентного РС и системных аутоиммунных заболеваний.
Характерной особенностью РС является наличие интратекального синтеза IgG в виде повышенного индекса IgG или олигоклональных групп IgG в ликворе, выявляемое более чем в 85% случаев. При AQP4+ РСОНМ и MOG-АР олигоклональные группы IgG обнаруживаются только в 10–20% случаев и могут исчезать во время фазы ремиссии, при саркоидоз-ассоциированной миелопатии — в 23% [20].
Плеоцитоз ликвора до 50 клеток в 1 мм3 с преобладанием лимфоцитов обычно наблюдается примерно в 50% случаев РС, 14–79% — AQP4+ РСОНМ и 50–75% — MOG-АР. Превышение этого значения отмечается в 13–35% случаев AQP4+ РСОНМ и 19% — MOG-АР (а выше 100 клеток в 1 мм3 — в 12%) и почти исключает диагноз «РС». При саркоидоз-ассоциированной миелопатии плеоцитоз обнаруживался у 79% пациентов и в среднем составлял 43 клетки (диапазон — 16–88) [18][20].
Для подтверждения диагноза иммуноопосредованного демиелинизирующего миелита необходимо проведение тестирования на соответствующие антитела. Определение MOG-антител рекомендуется проводить при клинической картине и данных нейровизуализации с типичными для MOG-АР признаками, но атипичными для РС. При этом предпочтительным образцом в настоящее время признана сыворотка крови, а рекомендуемым методом анализа — метод клеточной презентации антигена на живых клетках. Более высокая степень диагностической достоверности определяется более высоким титром MOG-антител (>1:100). В то же время одновременное обнаружение MOG-антител в сыворотке и в ликворе наблюдается у 41–87% пациентов, поэтому в подозрительных случаях при их отсутствии в сыворотке крови необходимо проводить тестирование ликвора. Ситуация с определением AQP4-антител аналогична. MOG- и AQP4-антитела являются наиболее часто выявляемыми и наиболее изученными, однако возможны и случаи двойного серонегативного заболевания спектра оптиконейромиелита, что предполагает наличие ещё не описанных антител. GFAP-антитела необходимо определять в ликворе, что имеет большую чувствительность и специфичность по сравнению с тестированием сыворотки [4][6][7][24–26].
При проведении дифференциальной диагностики воспалительных и невоспалительных миелопатий необходимо учитывать особенности клинических проявлений различных по этиологии синдромов поражения спинного мозга, возможность вовлечения экстраспинальных структур, особенности начала и дальнейшего развития патологического процесса. Особое внимание должно уделяться полноте проведения МРТ-исследования — нейровизуализации всех отделов спинного мозга, анализу как сагиттальных, так и аксиальных проекций с учётом известных паттернов накопления контрастного вещества. Решающее значение для определения этиологии спинального поражения имеет лабораторная диагностика.
1. Sechi E., Flanagan E.P. Evaluation and Management of Acute Myelopathy. Semin. Neurol. 2021; 41(05):511-529. https://doi.org/10.1055/s-0041-1733792
2. Pardo C.A. Clinical Approach to Myelopathy Diagnosis. Continuum (Minneap Minn). 2024;30(1):14-52. https://doi.org/10.1212/CON.0000000000001390
3. Valencia-Sanchez К., Flanagan E.P. Uncommon inflammatory/immunerelated myelopathies. J. of Neuroimmunology. 2021;361:577750. https://doi.org/10.1016/j.jneuroim.2021.577750
4. Jarius S., Aktas O., Ayzenberg I., Bellmann-Strobl J., Berthele A., Giglhuber K. et al. Update on the diagnosis and treatment of neuromyelits optica spectrum disorders (NMOSD) – revised recommendations of the Neuromyelitis Optica Study Group (NEMOS). Part I: Diagnosis and differential diagnosis. J. Neurol. 2023;270:3341-3368. https://doi.org/10.1007/s00415-023-11634-0
5. Евтушенко С.К., Скоромец А.А., Скоромец А.П., Евтушенко О.С. Избранные лекции по детской и общей неврологии. М.: Медицинское информационное агентство; 2021.
6. Cacciaguerra L., Flanagan E.P. Updates in NMOSD and MOGAD Diagnosis and Treatment: A Tale of Two Central Nervous System Autoimmune Inflammatory Disorders. Neurologic Clinics. 2024;42(1):77-114. https://doi.org/10.1016/j.ncl.2023.06.009
7. Passeri M., Matthews E., Kammeyer R., Piquet A.L. Update in autoimmune and paraneoplastic myelopathies: Newly described antigen targets and antibody testing. Front. Neurol. 2022;13:972143. https://doi.org/10.3389/fneur.2022.972143
8. Скоромец А.А., Афанасьев В.В., Скоромец А.П., Скоромец Т.А. Сосудистые заболевания спинного мозга. СПб.: Политехника, 2019:400.
9. Zalewski N.L., Rabinstein A.A., Krecke K.N., Brown R.D.Jr., Wijdicks E.F.M., Weinshenker B.G. et al. Characteristics of Spontaneous Spinal Cord Infarction and Proposed Diagnostic Criteria. JAMA Neurol. 2019;76(1):56-63. https://doi.org/10.1001/jamaneurol.2018.2734
10. Murphy O.C., Barreras P., Villabona-Rueda A., Mealy M., Pardo C.A. Identification of specific causes of myelopathy in a large cohort of patients initially diagnosed with transverse myelitis. J. of the Neurological Sciences. 2022;442:120425. https://doi.org/10.1016/j.jns.2022.120425
11. Cacciaguerra L., Sechi E., Rocca M.A., Filippi M., Pittock S.J., Flanagan E.P. Neuroimaging features in inflammatory myelopathies: A review. Front. Neurol. 2022;13:993645. https://doi.org/10.3389/fneur.2022.993645
12. Barreras P., Fitzgerald K.C., Mealy M.A., Jimenez J.A., Becker D., Newsome S.D. et al. Clinical biomarkers differentiate myelitis from vascular and other causes of myelopathy. Neurology. 2018;90:e12-21. https://doi.org/10.1212/WNL.0000000000004765
13. Levy M. Immune-Mediated Myelopathies. Continuum (Minneap Minn). 2024;30(1):180-198. https://doi.org/10.1212/CON.0000000000001382
14. Lopez Chiriboga S., Flanagan E.P. Myelitis and other autoimmune myelopathies. Continuum (Minneap Minn). 2021;27(1):62-92. https://doi.org/10.1212/CON.0000000000000900
15. Pfausler B., Rass V., Lindner A. Infektionen des Rückenmarks und der angrenzenden Strukturen [Infections of the spinal cord and adjacent structures]. Nervenarzt. 2023;94(4):287-295. (In German). https://doi.org/10.1007/s00115-023-01439-x
16. Fletcher A.M., Bhattacharyya S. Infectious Myelopathies. Continuum (Minneap Minn). 2024;30(1):133-159. https://doi.org/10.1212/CON.0000000000001393
17. Marignier R. Acute and subacute myelopathy. Revue Neurologique. 2021;177(5):557-566. https://doi.org/10.1016/j.neurol.2020.08.003
18. Fadda G., Flanagan E.P., Cacciaguerra L., Jitprapaikulsan J., Solla P., Zara P., Sechi E. Myelitis features and outcomes in CNS demyelinating disorders: Comparison between multiple sclerosis, MOGAD, and AQP4-IgG-positive NMOSD. Front. Neurol. 2022;13:1011579. https://doi.org/10.3389/fneur.2022.1011579
19. Пономарев Г.В., Скоромец А.А., Краснов В.С., Родионова О.В., Глистенкова Д.Д., Порхун Н.Ф., и др. Сосудистая миелопатия: причины и механизмы, возможности диагностики и лечения. Неврология, нейропсихиатрия, психосоматика. 2018;10(1):12-16.
20. Murphy O.C., Salazar-Camelo A., Jimenez J.A., Barreras P., Reyes M.I., Garcia M.A. et al. Clinical and MRI phenotypes of sarcoidosis-associated myelopathy. Neurol. Neuroimmunol. Neuroinflamm. 2020;7:e722. https://doi.org/10.1212/NXI.0000000000000722
21. Fink J.K. Hereditary Myelopathies. Continuum (Minneap Minn). 2021;27(1):185-204. https://doi.org/10.1212/con.0000000000000934
22. Kranz P.G., Amrhein T.J. Imaging approach to myelopathy: acute, subacute, and chronic. Radiol. Clin. North. Am. 2019;57:257-279. https://doi.org/10.1016/j.rcl.2018.09.006
23. Sechi E., Morris P.P., McKeon A., Pittock S.J., Hinson S.R., Weinshenker B.G. et al. Glial fibrillary acidic protein IgG related myelitis: characterisation and comparison with aquaporin-4-IgG myelitis. J. Neurol. Neurosurg. Psychiatry. 2019;90:488-490. https://doi.org/10.1136/jnnp-2018-318004
24. Banwell B., Bennett J.L., Marignier R., Kim H.Jin., Brilot F., Flanagan E.P. et al. Diagnosis of myelin oligodendrocyte glycoprotein antibody-associated disease: International MOGAD Panel proposed criteria. Lancet. Neurol. 2023:22(3);268-282. https://doi.org/10.1016/S1474-4422(22)00431-8
25. Saballegue J.D., Tiongson M.P. Rituximab Therapy for Double Seronegative Neuromyelitis Optica Spectrum Disease. Cureus 2024;16(5):e60004. https://doi.org/10.7759/cureus.60004
26. Zara P., Dinoto A., Carta S., Floris V., Turilli D., Budhram A. et al. Non-demyelinating disorders mimicking and misdiagnosed as NMOSD: a literature review. Eur. J. Neurol. 2023;30:3367-3376. https://doi.org/10.1111/ene.15983
Сорокин Юрий Николаевич, д.м.н., доцент; профессор кафедры неврологии и восстановительной медицины с курсом остеопатии
Ростов-на-Дону
Сорокина Елена Юрьевна, старший преподаватель кафедры иностранных языков с курсом латинского языка
Ростов-на-Дону
Сорокин Ю.Н., Сорокина Е.Ю. Особенности дифференциальной диагностики воспалительных и невоспалительных миелопатий. Южно-Российский журнал терапевтической практики. 2025;6(2):118-128. https://doi.org/10.21886/2712-8156-2025-6-2-118-128
Sorokin Yu.N., Sorokina E.Yu. Peculiarities of differential diagnostics of inflammatory and noninflammatory myelopathies. South Russian Journal of Therapeutic Practice. 2025;6(2):118-128. (In Russ.) https://doi.org/10.21886/2712-8156-2025-6-2-118-128
344022, г. Ростов-на-Дону, пер. Нахичеванский, 29
tel.: +79185710558
e-mail: therapeuticjour@gmail.com (sylanadel@gmail.com)






















